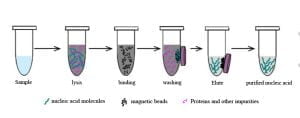
ขั้นตอน qPCR สำหรับการตรวจจับ Chlamydia เกี่ยวข้องกับสองขั้นตอนหลัก: การสกัดดีเอ็นเอและการขยายและการตรวจสอบ DNA ที่ตามมา. เริ่มแรก, เราจะมุ่งเน้นไปที่การแบ่งปันเนื้อหาการเรียนรู้ที่เกี่ยวข้องกับการสกัดดีเอ็นเอ. บทความนี้นำไปสู่รากฐานทางทฤษฎีและการแนะนำสารรีเอเจนต์ที่ใช้.
หลักการทำให้บริสุทธิ์ทางทฤษฎีของการสกัดดีเอ็นเอก่อนทำการทดลอง, การทำความเข้าใจหลักการพื้นฐานและขั้นตอนการดำเนินงานเป็นประโยชน์. ช่วยให้เราเข้าใจการทดลอง, มองเห็นผลลัพธ์ที่เป็นไปได้, และหลีกเลี่ยงเพียงแค่เป็นผู้ให้บริการซ้ำ ๆ.
หลักการพื้นฐานของการสกัดดีเอ็นเอ
ความสมบูรณ์ของโครงสร้างดีเอ็นเอ: เช่น, หลักการที่อยู่เบื้องหลังการตรวจจับ Chlamydia ที่ใช้การตรวจสอบ qPCR เกี่ยวข้องกับการตรวจจับเฉพาะของหนองในเทียมโดยการขยายภูมิภาคการเข้ารหัส 16SRRNA ในจีโนม Chlamydia. การขยายเฉพาะของ Chlamydia สามารถตรวจพบได้ที่ 520nm (ช่อง FAM). DNA Chlamydia ที่ไม่สมบูรณ์จะล้มเหลวในการผ่านการขยายและตรวจจับลักษณะเฉพาะ.
การพิจารณา:
(1) เพื่อให้แน่ใจว่าสิ่งนี้, ขั้นตอนการทดลองของเราควรเกี่ยวข้องกับการจัดการอย่างอ่อนโยนเพื่อป้องกันการย่อยสลายของดีเอ็นเอ.
(2) ควรให้ความสนใจกับการยับยั้ง DNases.
(3) หากเราซื้อชุดรีเอเจนต์ QPCR Chlamydia เท่านั้นโดยไม่ต้องใช้ชุดรีเอเจนต์การสกัดที่สอดคล้องกัน, เราไม่เพียง แต่ต้องทดสอบการบังคับใช้ของชุดทดสอบ แต่ยังตรวจสอบประสิทธิภาพของกระบวนการสกัดของเรา.
1.1.2 ความบริสุทธิ์ของ DNA: พยายามกำจัดสารโมเลกุลขนาดใหญ่อื่น ๆ ที่อาจรบกวน DNA, ดังนั้นการหลีกเลี่ยงผลกระทบใด ๆ ต่อการทดลองที่ตามมา. ตรวจสอบให้แน่ใจว่าตัวอย่างที่สกัดไม่มีตัวทำละลายอินทรีย์หรือไอออนโลหะที่มีความเข้มข้นสูงที่ยับยั้งเอนไซม์.
การพิจารณา:
(1) ข้อควรพิจารณาที่คล้ายกันใช้เมื่อ จำกัด จำนวนเซลล์ทั้งหมดในตัวอย่างเซลล์, เนื่องจาก DNA ที่ไม่ใช่เป้าหมายภายในสารโมเลกุลขนาดใหญ่อื่น ๆ อาจส่งผลต่อการทดลอง.
(2) หมายเหตุเตือนสำหรับการควบคุมคุณภาพ (QC): การสุ่มตัวอย่างไม่ควรมากเกินไป. ในขณะที่การเพิ่มปริมาณการทดสอบอาจเป็นที่ต้องการสำหรับการเป็นตัวแทนใน QC, ควรใช้ความระมัดระวังในกรณีดังกล่าว.
(3) สิ่งสำคัญคือต้องเข้าใจตัวอย่างของเรา; หากมีตัวทำละลายอินทรีย์หรือไอออนโลหะที่ยับยั้งเอนไซม์, เราจำเป็นต้องยืนยันว่าสารเหล่านี้สามารถกำจัดได้อย่างมีประสิทธิภาพก่อนการทดลองหรือไม่หรือหากความเข้มข้นของพวกเขาสามารถลดลงได้ด้วยวิธีการเพาะเลี้ยงเซลล์.
(4) การปนเปื้อนจากกรดนิวคลีอิกอื่น ๆ เป็นปัจจัยรบกวนที่พบบ่อย.
โดยเฉพาะ, เมื่อจัดการ Chlamydia หรือ Chlamydia ตัวอย่างที่เป็นบวก vortexing, อาจมีการสร้างสเปรย์. เพราะฉะนั้น, ในระหว่างการสอบ Chlamydia, การดำเนินการปลอดเชื้ออย่างเข้มงวดภายในตู้ความปลอดภัยทางชีวภาพเป็นสิ่งสำคัญ, และการหมุนเหวี่ยงอย่างรวดเร็วหลังจากแนะนำให้ใช้กระแสน้ำวน.